Raising awareness for rare liver diseases: challenges and breakthroughs
Rare Disease Day, held on 28 February 2025, shines a light on challenges faced by over 300 million people worldwide living with rare diseases. For the hepatology community, this day is a reminder that continued research, increased awareness, and improved care for rare liver conditions is still needed. By enhancing collaboration among clinicians, researchers, and patient advocates, we can advance our understanding and develop better treatments, ensuring no patient is left behind in the fight against rare diseases.
Rare liver diseases: an overview
A disease is classified as rare when it affects no more than five individuals per 10,000 people. Globally, an estimated 6,000 to 8,000 rare diseases have been identified. Although each rare disease affects a small number of people, the vast number of these conditions means that, collectively, they are likely to impact around 7% of the global population. This means that in the European Union (EU) between 27 and 36 million people live with a rare disease. The European Liver Patients' Association (ELPA) estimates that rare liver diseases occur in approximately 1 out of every 50,000 to 100,000 births across all regions, but the precise prevalence remains uncertain. Rare liver diseases have a significant impact on patients’ lives, often leading to complex diagnostic and treatment challenges. The following table presents a list of rare liver diseases, excluding rare liver cancers, along with their commonly used abbreviations and estimated worldwide prevalence rates.
| Disease | Abbreviation | Prevalence |
| Acute Hepatic Porphyria | AHP | 1/75,000 |
| Alagille Syndrome | AS | 1/70,000 |
| Alpha-1 Antitrypsin Deficiency | AATD | 1-5/10,000 |
| Autoimmune Hepatitis | AIH | 1-5/10,000 |
| Biliary Atresia | BA | 1-9/100,000 |
| Budd-Chiari Syndrome | BCS | 1-9/100,000 |
| Cholesteryl Ester Storage Disease | CESD | NA |
| Congenital Hepatic Fibrosis | CHF | NA |
| Crigler-Najjar Syndrome | CNS | 1-9/100,000 |
| Galactosemia | - | NA |
| Glycogen Storage Diseases | GSDs | NA |
| Hereditary Hemochromatosis | HH | NA |
| Lysosomal Acid Lipase Deficiency | LAL-D | 1/177,000 |
| North American Indian Childhood Cirrhosis | NAIC | < 1/1,000,000 |
| Primary Biliary Cholangitis | PBC | 1-5/10,000 |
| Primary Sclerosing Cholangitis | PSC | 1-9/100,000 |
| Progressive Familial Intrahepatic Cholestasis | PFIC | 1/50,000–100,000 |
| Tyrosinemia Type 1 | HT-1 | 1/100,000 |
| Wilson’s Disease | WD | 1/30,000–110,000 |
*according to the Orphanet database; rare liver cancers not included.
Due to their low prevalence, these diseases are frequently under- or misdiagnosed, resulting in delayed treatment and poorer health outcomes. Increasing awareness within the medical community and advancing research are crucial for improving early detection, developing new therapies, and enhancing overall patient care.
Challenges in research and treatment
One of the primary challenges in addressing rare liver diseases is the small patient population, which makes it difficult to conduct large-scale clinical trials and gather comprehensive data. This limited pool of participants often leads to slower progress in understanding disease mechanisms and developing effective treatments. Additionally, funding limitations pose a significant barrier, as rare diseases may not attract the same level of financial support and investment as more common conditions. These challenges underscore the importance of international collaboration and the need for dedicated resources to drive innovation in this field.
The EU is actively working to improve access to diagnosis, care, and information through initiatives like the European Reference Networks (ERNs), research funding (Horizon Europe), digital databases (Orphanet) and platforms like the European Platform on Rare Disease Registration. The ERN Rare-Liver network, dedicated specifically to rare liver diseases, aims “to improve the care of rare liver disease patients throughout Europe”. Other collaborative projects and strategic partnerships, such as the European Rare Diseases Research Alliance (ERDERA), continue to stimulate advancements in the diagnosis and treatment for rare disease patients across Europe, while also supporting the alignment of rare disease strategies among countries and regions.
Latest clinical trials and innovations
Recent advancements in clinical research offer hope for those affected by rare liver diseases.
PBC
In September 2024, elafibranor was approved by the European Commission as treatment for PBC in combination with ursodeoxycholic acid (UDCA). Elafibranor is a peroxisome proliferator-activated receptor alpha/delta (PPAR-α/δ) agonist: by activating PPAR-α and PPAR-δ, it improves bile flow, reduces bile duct inflammation, and prevents liver damage, making it a promising treatment for PBC. Moreover, elafibranor is currently being investigated as treatment for patients with PSC.
On 16 January 2025, the Medicines and Healthcare products Regulatory Agency (MHRA) in the UK announced the approval of seladelpar, a PPAR-δ agonist, for the treatment of PBC in adults, later granted with a conditional marketing authorization by the European Commission on 20 February 2025. Seladelpar reduces liver inflammation and is prescribed alongside UDCA for patients with an inadequate response to UDCA or as a monotherapy in patients intolerant to UDCA. The recommended dosage of 10 mg once daily was shown to be efficacious in a 12-month clinical trial. Patients and healthcare providers are encouraged to report any side effects through the MHRA Yellow Card scheme, ensuring ongoing monitoring of the medication's safety and effectiveness.
Wilson’s Disease
An ongoing phase I/II clinical trial on gene therapy for Wilson’s Disease aims to evaluate the safety, tolerability, and pharmacological effects of VTX-801 over a period of five years. VTX-801 – a replication-deficient recombinant adeno-associated viral vector (rAAV) composed of an AAV liver tropic capsid – is designed to deliver an ssDNA of a functional ATP7B minigene (a shortened but effective version of the defective gene responsible for Wilson Disease). The trial aims to test up to three different therapeutic doses administered as a single intravenous dose in order to determine the safest and most effective therapy. Following the five-year follow-up, the study aims to determine whether VTX-801 can restore proper copper metabolism, potentially reducing or eliminating the need for lifelong medication in Wilson’s Disease patients.
Alpha-1 Antitrypsin Deficiency
A promising therapy for Alpha-1 Antitrypsin Deficiency involves reducing the production of the faulty AAT protein by silencing the SERPINA1 gene using RNA interference (RNAi). This leads to reduced production of misfolded alpha-1 antitrypsin protein (Z-AAT) preventing its accumulation in the liver and slowing the progression of liver disease in AATD patients. One such RNAi candidate showing promise in clinical studies is fazirsiran. Phase 2 studies have demonstrated that fazirsiran effectively lowers Z-AAT levels, potentially halting disease progression and allowing for liver regeneration. These encouraging results have led to the initiation of Phase 3 trials to further evaluate its efficacy and safety.
Another promising avenue being explored is gene therapy. One example is BEAM-302, a liver-targeting lipid-nanoparticle base editor that corrects the PiZ mutation in the SERPINA1 gene – the most common cause of severe AATD. By precisely editing this mutation, BEAM-302 aims to reduce the accumulation of misfolded AAT protein in the liver and increase the levels of functional AAT protein in circulation, thereby addressing both hepatic and pulmonary aspects of the disease. Unlike current therapies, it restores the gene’s natural function, enabling a proper inflammatory response. Preclinical studies suggest long-lasting effects, offering a potential durable solution for AATD. In a Phase 1/2 clinical trial in June 2024, a patient received a dose for the first time, marking a significant milestone in the development of this therapy.
| RNAi | RNA interference is a process where RNA molecules regulate gene expression in a sequence-specific manner by using double-stranded RNA to inhibit translation or transcription. |
| PiZZ | Severe form of alpha-1 antitrypsin deficiency where a person inherits two copies of the Z variant of the SERPINA1 gene, leading to low AAT levels and increased risk of lung and liver disease. |
| Base editor | Genetic tool that allows scientists to precisely change a single base in the DNA without cutting the DNA strand. |
The role of EASL and the future of rare liver disease research
EASL plays a key role in advancing the understanding and treatment of rare liver diseases. EASL provides comprehensive Clinical Practice Guidelines (CPGs) to support healthcare professionals in diagnosing and managing these complex conditions. Key guidelines include:
- EASL Clinical Practice Guidelines on genetic cholestatic liver diseases (2024)
- EASL Clinical Practice Guidelines on the management of cystic liver diseases (2022)
- EASL Clinical Practice Guidelines on sclerosing cholangitis (2022)
- EASL Clinical Practice Guidelines on haemochromatosis (2022)
- Role of endoscopy in primary sclerosing cholangitis: European Society of Gastrointestinal Endoscopy (ESGE) and European Association for the Study of the Liver (EASL) Clinical Guideline (2017)
- Stay tuned for new CPGs that will be released in 2025, which will update the EASL Clinical Practice Guidelines: Autoimmune hepatitis (2015) and the EASL Clinical Practice Guidelines on Wilson's disease (2012).
- An update of the EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis (2017) is in progress.
Two of these CPGs are already available in the EASL Guidelines App! Download it now to consult EASL Clinical Practice Guidelines anytime, anywhere.

The most recent EASL CPGs on genetic cholestatic liver diseases have an interesting section on the management of pruritus. Pruritus management is complex and often represents one of the most debilitating symptoms associated with many genetic cholestatic liver diseases. To address this, the EASL CPGs include a comprehensive treatment algorithm applicable to all cholestatic liver diseases, recommending separate approaches for children and adults (Figure 1). Moreover, these CPGs emphasise how difficult it is to conduct large-scale clinical trials and collect comprehensive data. The lack of high-quality studies on patients results in many recommendations being based on expert opinion and the consensus of the CPG committee.
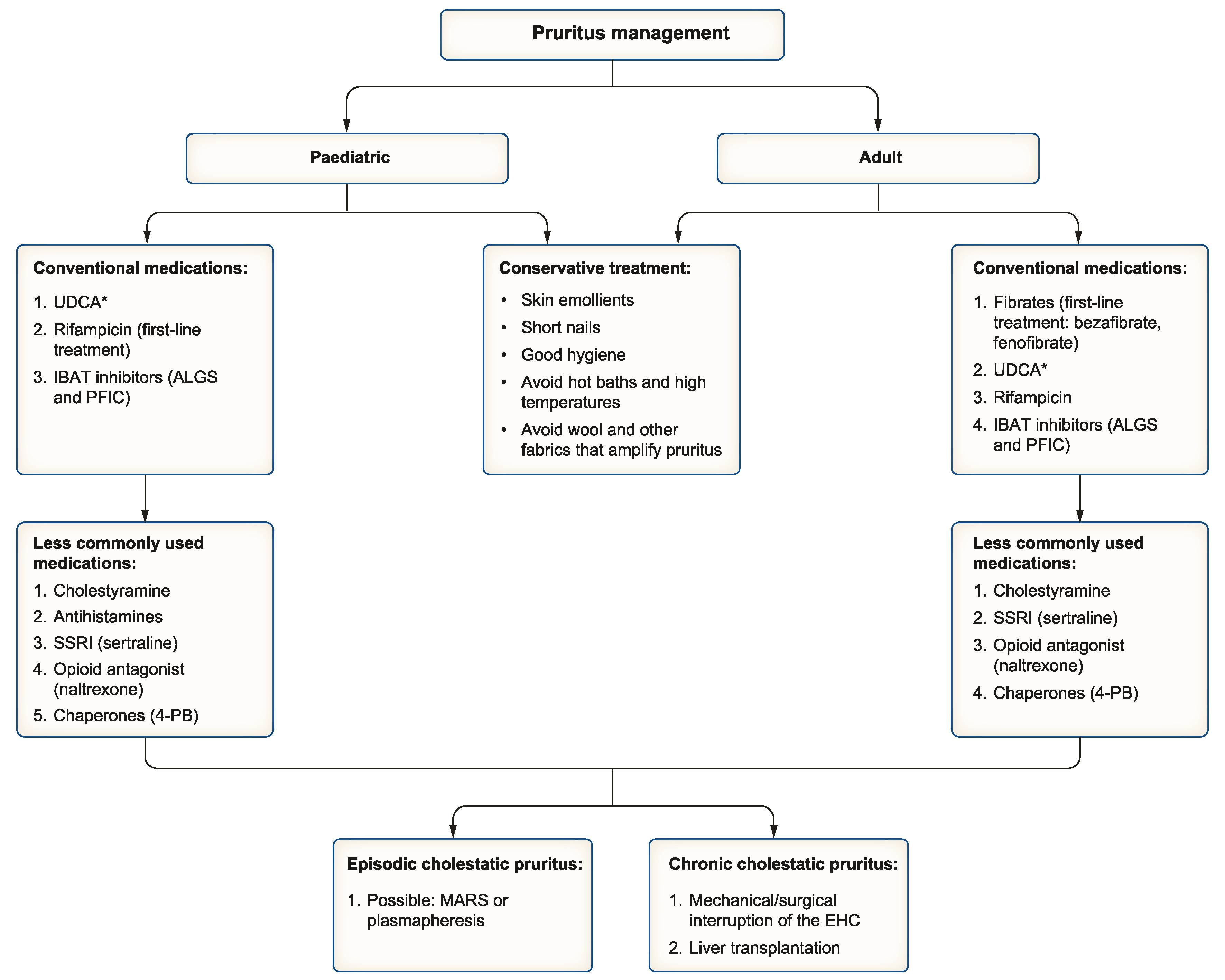
Figure 1: Pruritus management flowchart. ∗ UDCA is not generally considered a first-line treatment due to lack of evidence, however, because of its low risk profile, it is often tried as one of the first options in the management of cholestatic pruritus. ALGS, Alagille syndrome; EHC, enterohepatic circulation; IBAT, ileal bile acid transporter; MARS, molecular adsorbent recirculating system; PFIC, progressive familial intrahepatic cholestasis; SSRI, selective serotonin reuptake inhibitor; UDCA, ursodeoxycholic acid; 4-PB, 4-phenylbutyric acid.
Looking ahead, EASL remains committed to fostering research, improving clinical practice, and advocating for patients with rare liver diseases. By supporting multidisciplinary collaborations and promoting patient-centred care, EASL aims to drive future breakthroughs and ensure better outcomes for those affected by these challenging conditions.
Make your impact
Awareness is the first step toward change in the lives of those affected by rare liver diseases. You can make a difference by spreading the word, engaging in discussions, continuously learning and supporting research initiatives that will help improve patient outcomes. You can click here to find downloadable materials from the official Rare Disease Day 2025 website.
You can find below all EASL resources and events.
Resources
• EASL Quiz: 3 questions on Rare Liver Diseases and 12 questions on Immune-mediated & Cholestatic Diseases.
• ERN Rare-Liver: 29 on-demand webinars.
• EASL DeepDive: 2 episodes entitled “Understanding and managing PBC symptoms” and “Evolving insights in role and management of Alpha-1 Antitrypsin Deficiency”.
• EASL Genetic Grand Round: “Decoding cholestasis: genetic insights and case studies”.
• EASL Studio: “New treatment options for hereditary cholestasis syndromes: A game-changer?”, “EASL Haemochromatosis Guidelines 2022”, “Primary biliary cholangitis: A look at the future landscape of therapy for patients”, “Cholestatic pruritus: Can the itch be ditched?”, “Diagnostic and therapeutic challenges in Wilson’s disease”, “PBC: The itch we need to scratch”, “PSC and AIH overlap syndrome: does it actually exist?”, “Alpha-1-antitrypsin deficiency (AATD): A poster child for genetic therapy”, “What does the future management of PBC look like?”.
• EASL Studio Live at EASL Congress: “New therapeutic options and remaining challenges in PBC” (2024), “Challenges and opportunities for patients with PBC: Highlights from EASL Congress 2023” (2023), “Clinical trials in rare diseases – opportunities and challenges” (2023).
• EASL Policy Dialogues: “Rare but not forgotten – The challenges of living with PBC”, “Rarity and parity – Addressing inequities in rare liver disease”, “Enhancing treatment adherence & quality of life in rare liver disease”.
Upcoming events
February 2025
EASL DeepDive “What is the future of PBC treatment?” on 25 February 2025, 18:00 CET: register now!
March 2025
EASL Studio Season 8, Episode 5 “Lessons from Your PET: Understanding and Evaluating Treatment for Wilson’s Disease” on 5 March 2025, 18:00 CET.
EASL Studio Season 8, Episode 8 “Rare Liver Malignancies: Small Numbers, Big Questions” on 26 March 2025, 18:00 CET.
May 2025 - EASL Congress 2025, 7-10 May in Amsterdam
EASL Genetic Grand Round in-person (date TBD).
Wednesday, 7 May
Postgraduate Course: Mastering autoimmune and cholestatic liver disorders: Advanced clinical insights — Primary biliary cholangitis.
Postgraduate Course: Mastering autoimmune and cholestatic liver disorders: Advanced clinical insights — IgG4-related pancreatitis and cholangitis.
Postgraduate Course: Mastering autoimmune and cholestatic liver disorders: Advanced clinical insights — Autoimmune hepatitis.
Postgraduate Course: Mastering autoimmune and cholestatic liver disorders: Advanced clinical insights — Management of Primary sclerosing cholangitis.
State-of-the-art: Can genetics change our way of thinking about unexplained cholestasis?
Thursday, 8 May
EASL/IAIHG Do's and don'ts in immunosuppressive therapy in AIH.
Cholestasis of unknown origin.
Meet-the-Experts: Management of cholestatic itch: the role of IBAT inhibitors in clinical practice.
Friday, 9 May
EASL/ERN/ESPGHAN/ Do's and don'ts in diagnosis and management of Wilson's disease.
EASL - IPSCsg: Future of Hepatology: Should we target gut microbiota in cholestatic diseases?
Saturday, 10 May
Meet-the-Experts: Management of cystic liver diseases.
Summer 2025
In 2026, EASL will organise an EASL School entitled “Primary sclerosing cholangitis: from pathogenetic insights to novel therapies” in Hamburg, Germany. Stay tuned, applications will open this summer!
Articles and references
Rare Liver Diseases, American Liver Foundation, 2023.
Heneghan MA and Lohse AW. Update in Clinical Science: Autoimmune Hepatitis. J Hepatol. 2025 Jan 24:S0168-8278(24)02832-0. doi: 10.1016/j.jhep.2024.12.041.
Turner AM et al. Hepatic-targeted RNA interference provides robust and persistent knockdown of alpha-1 antitrypsin levels in ZZ patients. J Hepatol. 2018 Aug;69(2):378-384. doi: 10.1016/j.jhep.2018.03.012.
Strnad P et al. Fazirsiran for Liver Disease Associated with Alpha1-Antitrypsin Deficiency. N Engl J Med. 2022 Aug 11;387(6):514-524. doi: 10.1056/NEJMoa2205416. Epub 2022 Jun 25.
Remih K, Amzou S and Strnad P. Alpha1-antitrypsin deficiency: New therapies on the horizon. Curr Opin Pharmacol. 2021 Aug;59:149-156. doi: 10.1016/j.coph.2021.06.001. Epub 2021 Jul 10.
Plagiannakos CG et al. Treatment response and clinical event-free survival in autoimmune hepatitis: A Canadian multicentre cohort study. J Hepatol. 2024 Aug;81(2):227-237. doi: 10.1016/j.jhep.2024.03.021.
Leung KK et al. Primary Sclerosing Cholangitis-Inflammatory Bowel Disease: Epidemiology, Mortality, and Impact of Diagnostic Sequence. JHEP Reports. Article in press.
Di Giorgio A et al. Real-world experience with Odevixibat in children with Progressive Familial Intrahepatic Cholestasis. JHEP Reports. Article in press.
Wang C et al. Integrating electronic health records and GWAS summary statistics to predict the progression of autoimmune diseases from preclinical stages. Nat Commun. 2025 Jan 2;16(1):180. doi: 10.1038/s41467-024-55636-6.